Methods and applications of graph genomes to pathogens and public health
Michael B. Hall
Second Year Report
Presentation Overview
- What have I been doing for the last 12 months?
- Executive Summary
- Thesis Plan
- Publication Strategy
Presentation Overview
- What have I been doing for the last 12 months?
- Executive Summary
- Thesis Plan
- Publication Strategy
PhD-relevant time
- Refactoring/improving code for method in Chapter 1
- Re-implementing evaluation framework for Chapter 1
- Supporting collaborators
- Written methods for Chapter 1 paper
Other time
- Introduction to Container computing with Singularity
- Primers for predocs
bashcourse - Website for EMBL PhD Symposium
- Puntseq
- Conferences
- London Calling
- Applied Bioinformatics and Public Health Microbiology
- Computational Pan-genomics workshop
Presentation Overview
- What have I been doing for the last 12 months?
- Executive Summary
- Thesis Plan
- Publication Strategy
Aims of this PhD
Develop algorithms and software for variant discovery using bacterial genome graphs, building on work of a previous student in the lab.
Benchmark Nanopore versus Illumina SNP calling and show our algorithms meet the needs of clinical and public health users.
Improve upon current whole-genome sequencing-based drug resistance prediction for M. tuberculosis using genome graphs.
Curate a high-quality reference pan-genome for M. tuberculosis that includes a detailed map of the pe/ppe genes.
Presentation Overview
- What have I been doing for the last 12 months?
- Executive Summary
- Thesis Plan
- Publication Strategy
Chapter 1
Variant discovery in genome graphs
Develop algorithms and software for variant discovery using bacterial genome graphs, building on work of a previous student in the lab.
80%
Pan-genome
Bacterial genomes are incredibly diverse.
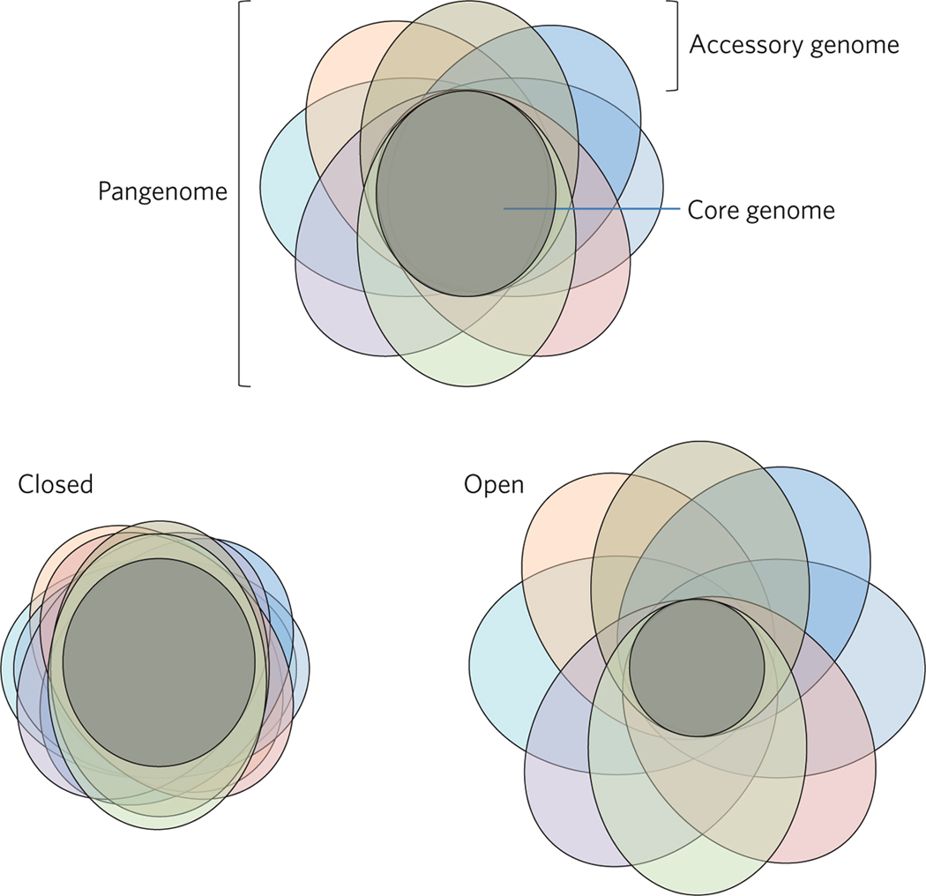
In an “open” pan-genome, such as Salmonella enterica, two individuals could share as little as 16% of their genes
The single-reference problem
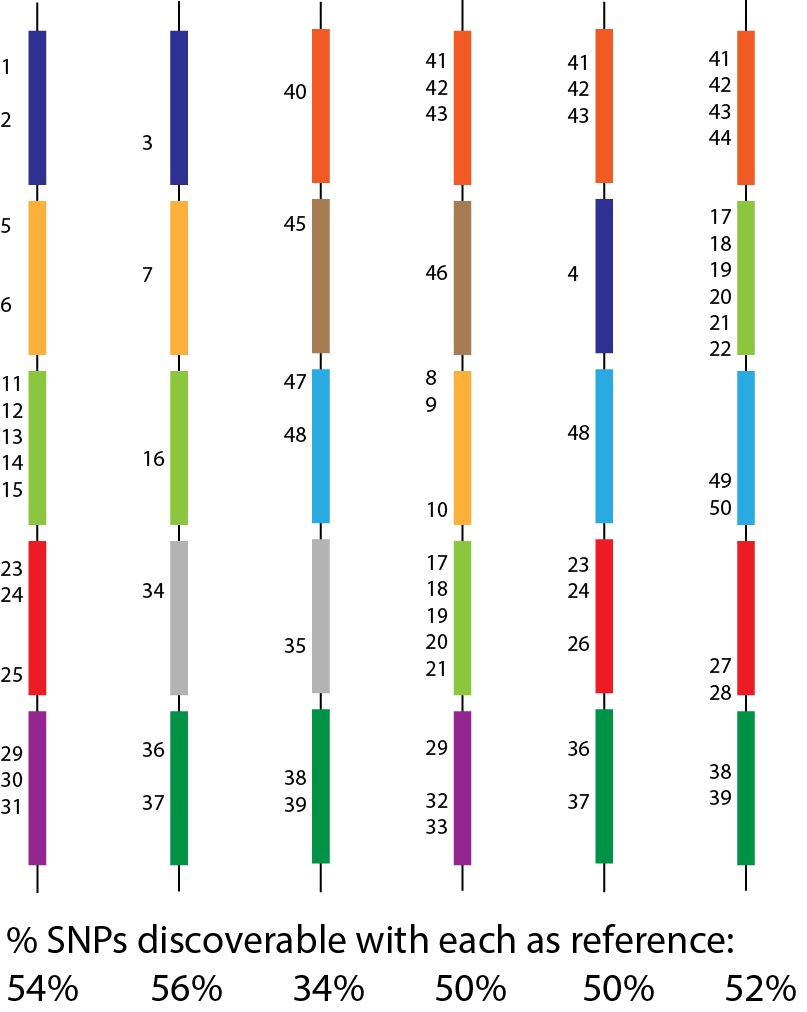
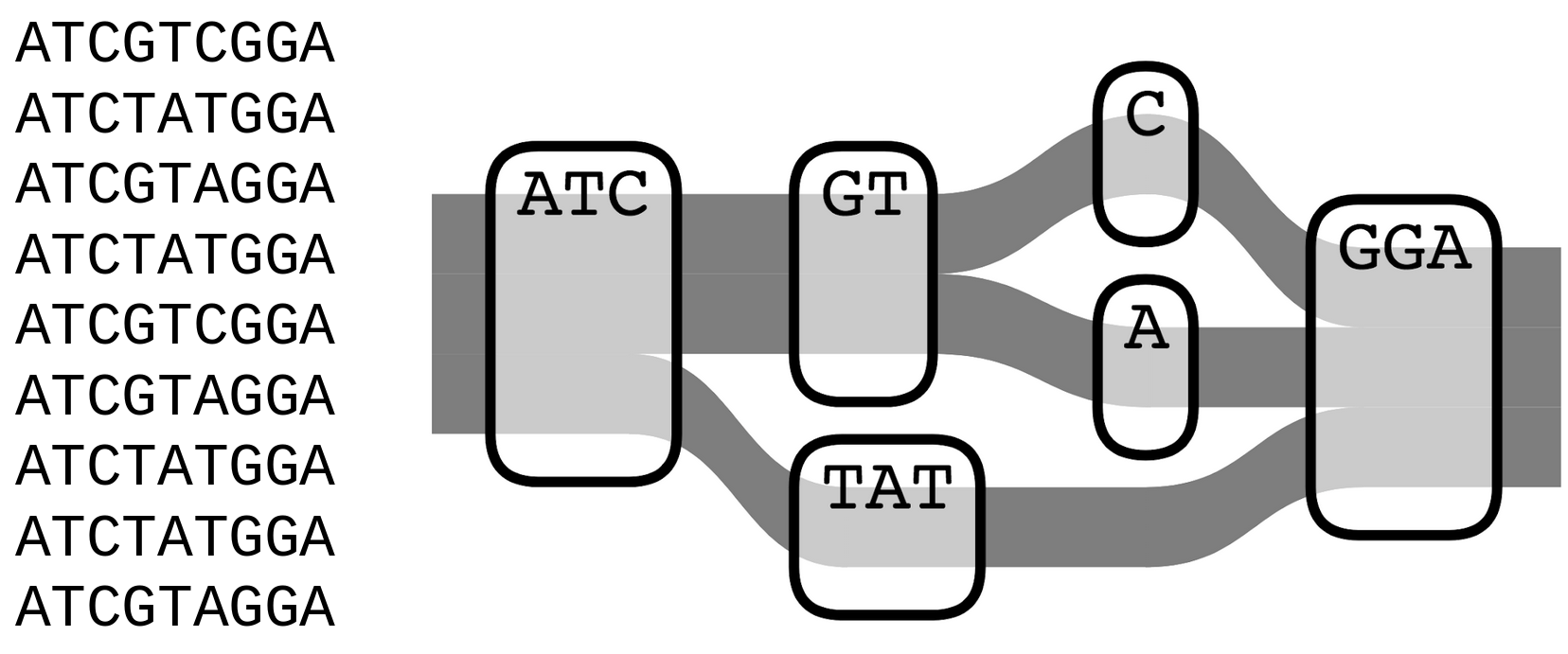
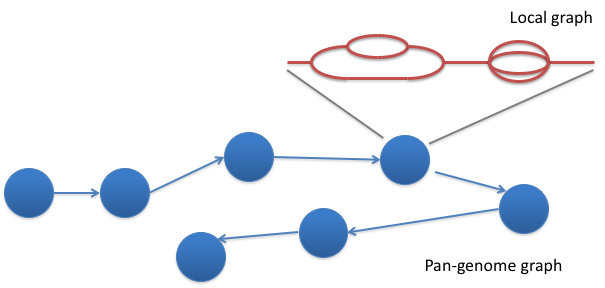
Genome graphs
- Uses a population reference graph (PRG) instead of a single, linear reference
- PRG represents variation seen within a population
- Two forms - local and pan-PRG
Nanopore sequencing
Variant calling in its infancy (medaka and nanopolish), but no extensive benchmark has been completed
Genome Graphs + Nanopore
Pandora - pan-genome inference and genotyping with long-noisy or short-accurate reads from Rachel Colquhoun.
Pandora map
Infer consensus sequence for a single sample and genotype with respect to this consensus sequence

Pandora compare
Infer consensus sequence for a collection of samples and genotype with respect to this consensus sequence
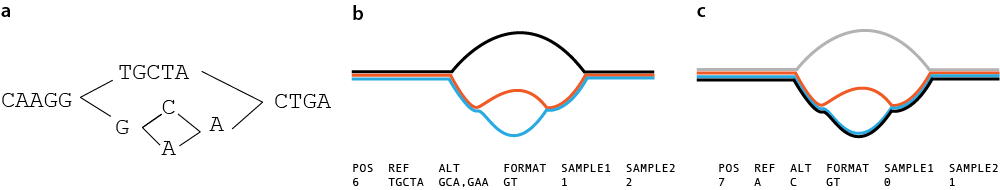
Limitation
Pandora can only genotype based on variation within the graph
Solution
The work in my first chapter outlines a method for removing this limitation and provides
an analysis of the gain in recall and precision by incorporating de novo variant discovery
into the pandora workflow.
What have I done?
-
Implement de novo variant discovery module within
pandora(~850/~3200 lines of source/test code) - Built evaluation framework (~1250/~3500 lines of source/test code)
-
Built a
snakemakepipeline of ~3500 lines of codes to orchestrate the entire evaluation and simulations.
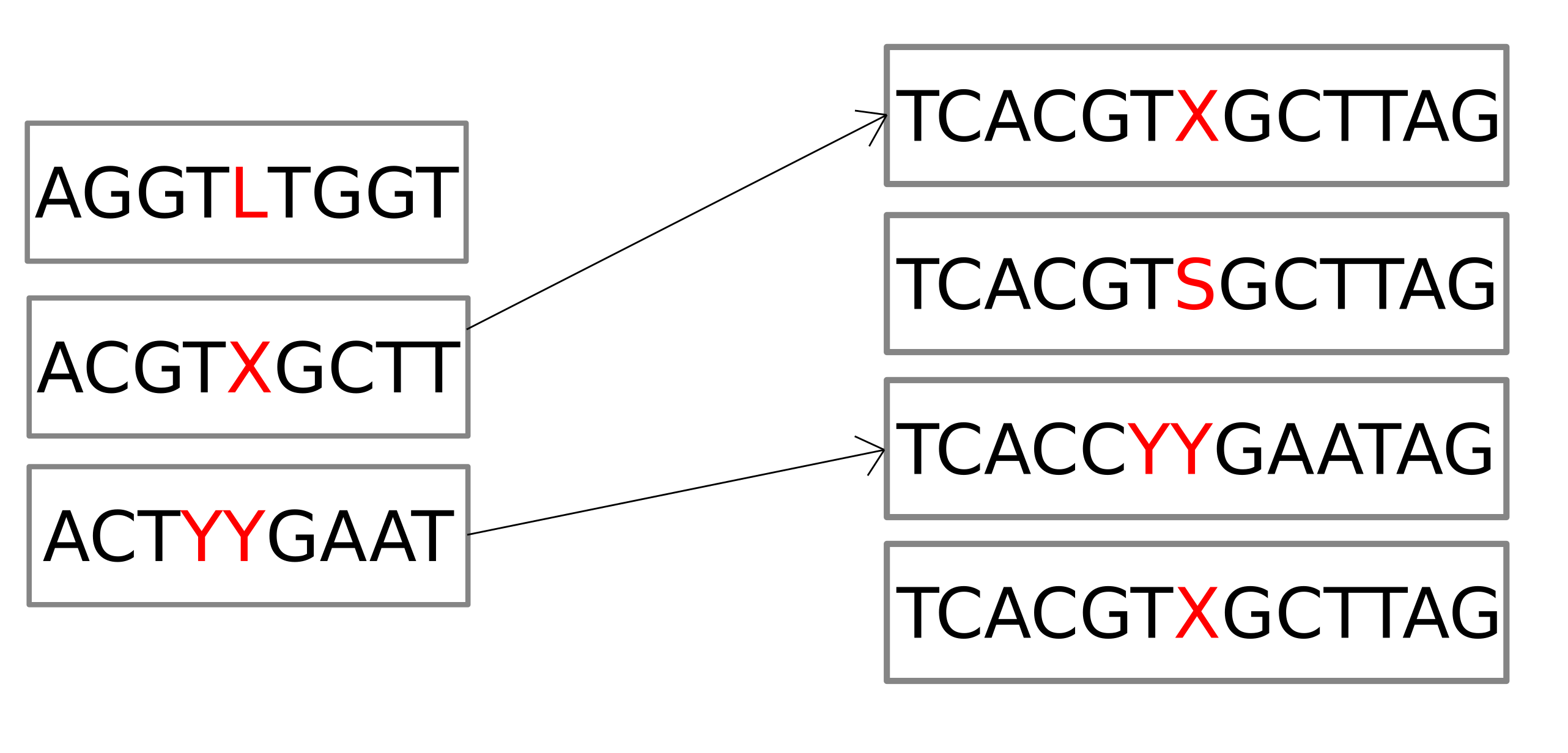
Variant discovery in a genome graph
Step 1: Finding candidate regions
Step 2: Enumerating paths through candidate regions
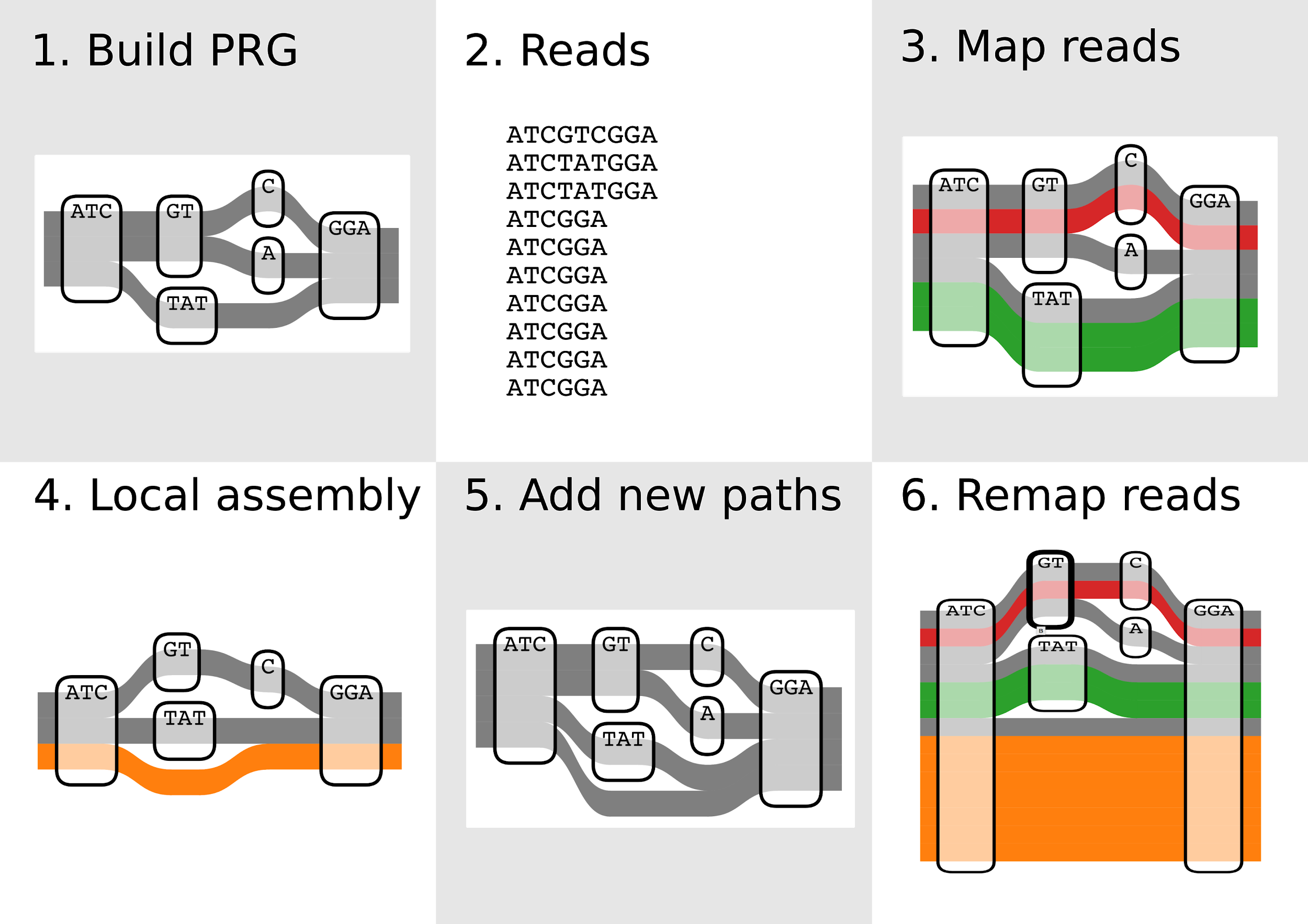
Evaluation
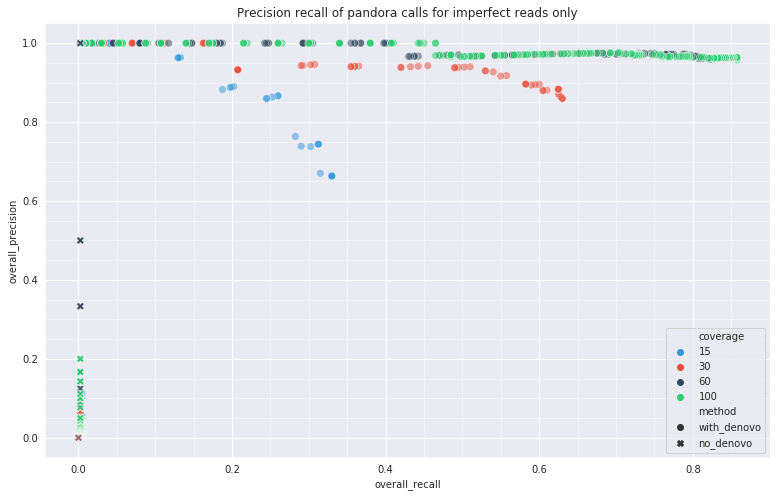
Simulated data
Aim to show that the addition of de novo discovery allows pandora to improve its
ability to discover and call variants correctly (precision/recall).
Simulated data
- 100 local PRGs (genes)
- Random path from each joined together into a genome
- Introduce variants at a given rate
- Simulate Nanopore reads from mutated genome
- Run
pandorawith reads from mutated genome - Assess how many introduced variants were found
Assessing variants
Simulation results
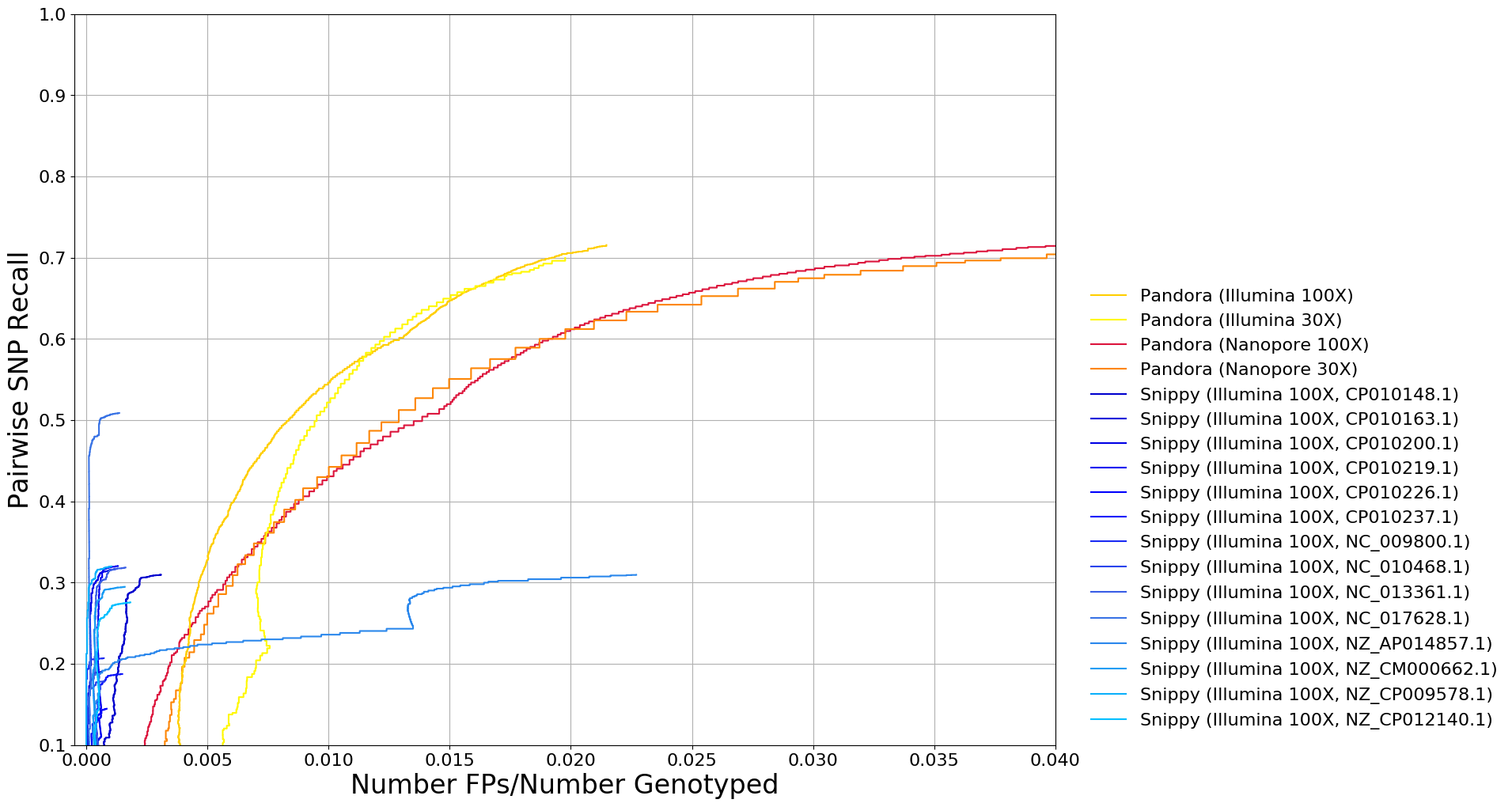
Empirical data
- The main focus of both
pandoraand de novo evaluation - Use
compareroutine to show the power of the reference-graph approach - 4 E. coli samples from different phylogroups
- Compare to other variant callers -
snippy,medaka,nanopolish- using a variety of references
Empirical data
Difficulty in evaluating four-way is “truth”
- Align each pair of genomes with
nucmerto get differences - Construct truth panel from these differences
- Map truth panel to a panel of probes from
pandoraVCF - Calculate recall and precision for all pairs
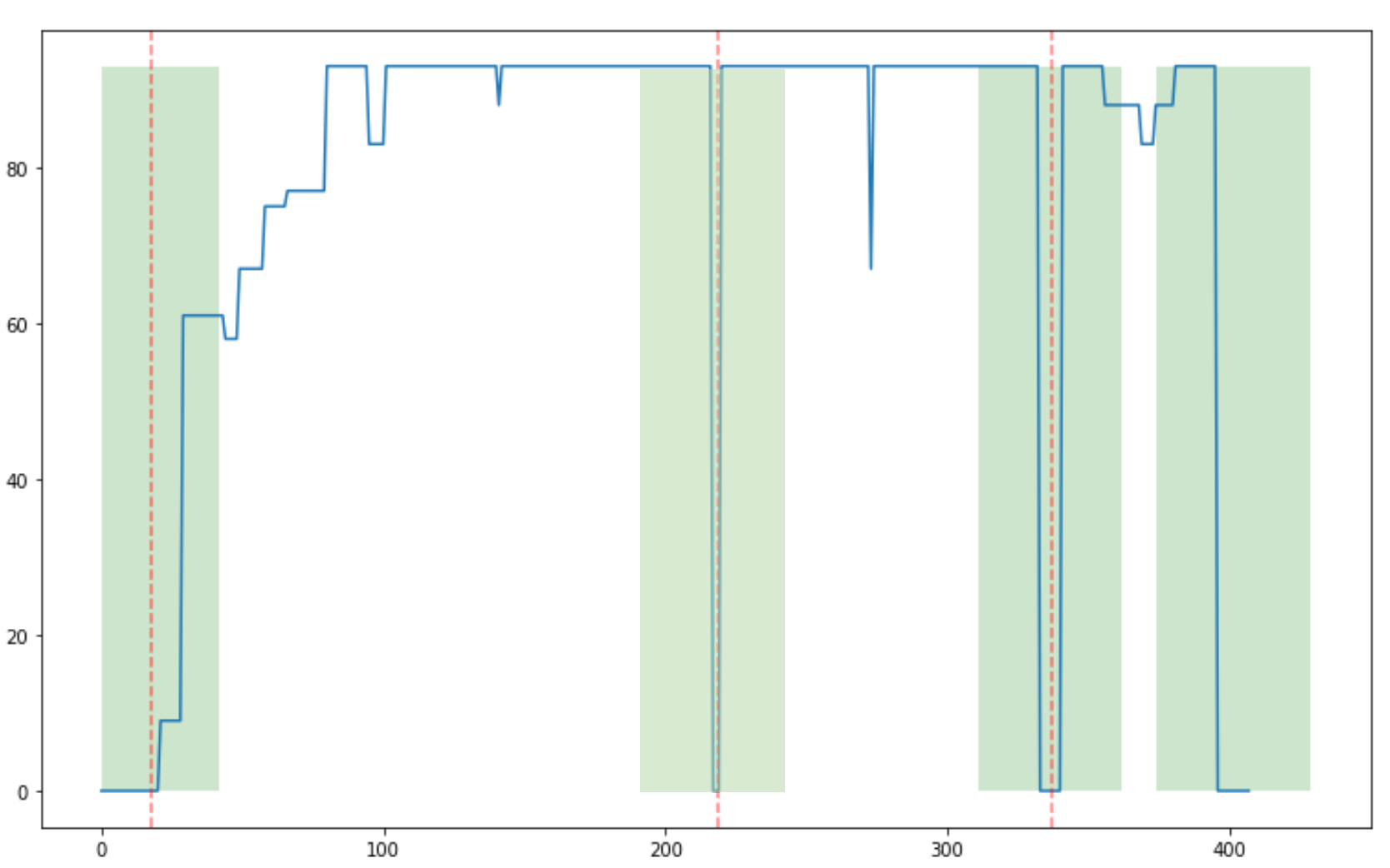
Preliminary Results
Outstanding work
- Completion of the four-way analysis
- Direct integration of de novo variants back into PRG
- 100-way analysis to validate the limit in variant detection using single-reference approaches
Chapter 2
Genome graph applications for M. tuberculosis and public health
Benchmark Nanopore versus Illumina SNP calling and show our algorithms meet the needs of clinical and public health users.
10%
Tuberculosis
- #1 cause of death by a single pathogen
- Standard of care requires phenotypic testing (DST) of the infecting organism - Mycobacterium tuberculosis - against the four first-line drugs
- M. tuberculosis is slow-growing - gold-standard DST takes ~2 months
TB and Public Health
- Public health requirements for TB diagnostics are resistance prediction, species identification, and clustering of genomes.
- Requirements currently met with Illumina
- Reasons to consider switching to Nanopore: cost, location of burden, speed
- Patient to result in 12.5 hours (2017) with Nanopore - and yield has improved since then
Genetic clustering
The first step towards clustering a set of genomes is determining a distance matrix.
Feeding aligned genomes into a phylogenetic tree-building tool- Counting SNP differences and clustering based on these
Genetic clustering
We define genetic distance to be the sum of genetic discordances, where missing data and heterozygosity do not cause discordance (unless the zygosity does not include the reference allele) and study the clustering this definition generates.
Chapter Aims
M. tuberculosis public health applications
Species identification- Prediction of drug resistance
- Epidemiological clustering of sample
How can pandora be used to improve these requirements?
Show that Nanopore sequencing is now capable of performing these tasks
Dataset
Same isolate DNA extraction sequenced on both Illumina and Nanopore
- 119 samples from Madagascar (35 PacBio)
- 83 samples from South Africa (evidence of transmission pairs)
- 20 samples from Birmingham
What have I done?
- Getting data from collaborators
- Organising data
- Repeat
- Providing assistance to collaborators
- Preliminary results of data quality and resistance-calling for collaborators
What’s left to do?
Analysis
Baseline variant analysis
Variant truth sets
- Illumina -
clockwork, combining best ofsamtoolsandcortex - Nanopore -
samtoolswith some filtering and masking
Baseline Illumina/Nanopore concordance, using PacBio as a validation (where we have it)
Variant Calling
Using four PRGs of varying complexity:
- Call SNPs and indels with
pandora - Compare to baseline calls
- Report concordance rate
- How does complexity of PRG affect concordance and computational cost
Clustering
- Calculate pairwise SNP distance from truth-set
- Calculate pairwise SNP distance from
pandorawith “best” PRG - Main figure will be dot plot of the two distance matrices - hoping for linear relationship
Chapter 3
Prediction of drug resistance in M. tuberculosis using genome graphs and Nanopore sequencing
Improve upon current whole-genome sequencing-based drug resistance prediction for M. tuberculosis using genome graphs.
Tuberculosis
- WGS-based diagnostics offer faster solution
- Two-week “liquid culture” gives similar results
- For the four first-line drugs, a study by the CRyPTIC consortium demonstrate that DST is not required if genotype predicts susceptibility
CRyPTIC
Comprehensive Resistance Prediction for Tuberculosis: an International Consortium
- Perform DST (14 drugs) and WGS on 40,000 global M. tuberculosis samples (many MDR)
- Combine with WGS data from another 60,000 samples
- The aim is to improve genotypic resistance prediction by expanding our catalogue of resistance mutations.
Mykrobe
Uses a panel of resistance markers to predict drug resistance from WGS data for M. tuberculosis and Staphylococcus aureus.
The predictive power of Mykrobe likely to expand during this PhD due to CRyPTIC consortium.
Chapter Aims
Nanopore concordance with Illumina for phenotype prediction in M. tuberculosis
Given the collection of SNPs and indels identified as being necessary for resistance to the 14 major drugs tested, we want to show that we can detect them as well with Nanopore data as we can with Illumina.
Limitations of existing methods
- Only two support Nanopore -
MykrobeandTBProfiler- small sample sizes (n<6) used to validate - The same panel produces different results between tools
TBProfileruses pileup approach - poor indel power. Indels are important for resistance to some drugsMykrobeuses k-mer mapping - requires high coverage. K-mers considered in isolation- Both only genotype wrt known variants
- CRyPTIC have shown flagging unknown mutations can lead to specificity and sensitivity acceptable for clinical usage (used by PHE)
Solution = Pandora
Can use smaller k-mer size than Mykrobe as it takes context into account. Therefore it theoretically requires less coverage.
Can call novel variants (Chapter 1)
Drug susceptibility prediction for M. tuberculosis using pandora
- Produce gene-succinct PRG of variants known to cause resistance/susceptibility
- Write a software program that takes
pandoraoutput and produces resistance predictions or flag for phenotyping - Validate on data from Chapter 2 against
Mykrobefor Illumina and Nanopore
Chapter 4
Construction of a M. tuberculosis reference pan-genome
Curate a high-quality reference pan-genome for M. tuberculosis that includes a detailed map of the pe/ppe genes.
Reasons for a M. tuberculosis pan-genome
- Closed pan-genome
- Some genes not present in H37Rv
- ~10% of the genome consists of pe/ppe genes
The enigmatic pe/ppe genes
- Implicated in immune evasion and virulence
- A disproportionately large amount of genetic diversity
- Nucleotide diversity ~2-fold higher than rest of the genome
- Sufficiently similar that short reads fail to map
- Frequently masked out of analyses
Chapter Aims
The ability to accurately map sequencing reads to these genes would likely improve our ability to perform variant calling in M. tuberculosis and therefore better determine how isolates relate to each other.
Build a high-quality pan-genome for M. tuberculosis, to allow variant discovery in all genes - ideally including the pe/ppe genes.
Data
- Assemble highest quality genomes from Chapter 2 plus very high-quality data from outside this thesis
- Assemblies will act as a “scaffold” for the pan-genome along with CRyPTIC variants
- Divide the genome into discrete pieces
Genome graph map of pe/ppe genes
- If pe/ppe genes arose via gene conversion short reads likely multi-map
- With high-quality short+long read assemblies, we hope to improve current resolution and allow more accurate mapping
Produce a collection of high-quality pe/ppe PRGs with information about what read length will provide reliable mapping, and whether Illumina data can be reliably mapped to them.
Analysis
Re-analyse data from Chapter 2 and see if we are better able to cluster samples with this new pan-genome with pe/ppe map
Assess variation in pe/ppe genes across 10,000 samples from CRyPTIC
Presentation Overview
- What have I been doing for the last 12 months?
- Executive Summary
- Thesis Plan
- Publication Strategy
Chapter 1
- The paper covering
pandoraand the work in Chapter 1 is currently in preparation - Rachel Colquhoun first-author and I will be the second author.
- The work I will have contributed to this paper includes the addition of the de novo variant discovery and a large amount of the evaluation of
pandora. - We aim to submit the paper by the end of 2019.
Chapter 2 and 3
- Combined into a single paper
- I will be the first author
- Aim to have work completed and manuscript submitted in the second quarter of 2020
Chapter 4
Too far off to say at this stage
Acknowledgements

